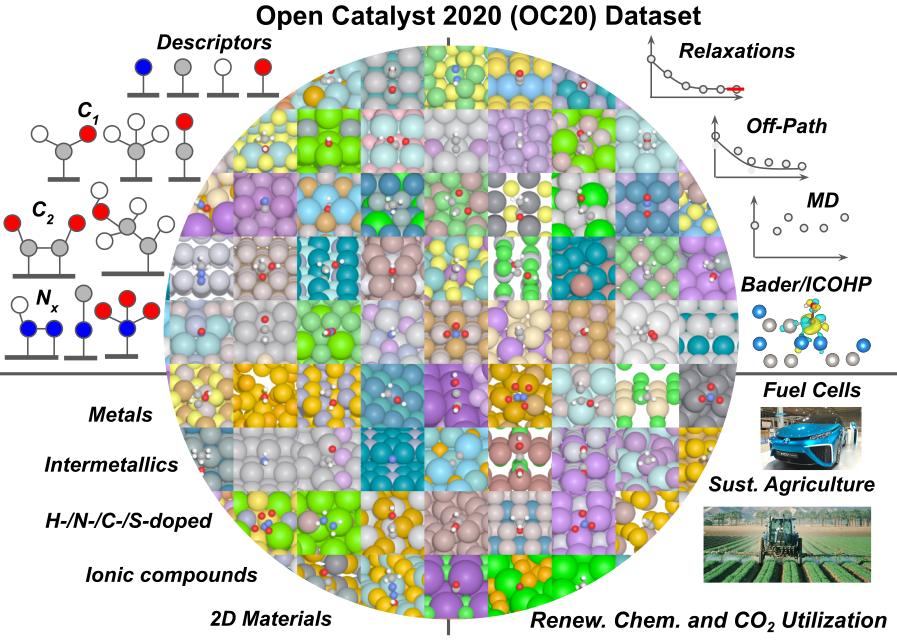
The Open Catalyst Project and OC20 Dataset
We are working with collaborators at Facebook AI Research on the Open Catalyst Project. We released the first step in this project - an open dataset of more than 1.2M adsorbate relaxations and 250M DFT single-point calculations across catalyst materials and chemistry space.
OCP Models
In addition to the dataset, we have also released a set of baseline models on github using state-of-the-art graph convolution models. These demonstrate what is feasible with current models and how to work and interact with the dataset. Get in touch if you have questions!
Press/PR
Software
We develop methods for machine learning in materials, uncertainty quantification, and automation. You can find a complete list of our software efforts on github
GASpy
Automating surface chemistry or catalysis calculations can be complicated. Our workflows are built on top of fireworks, pymatgen, luigi, ase, and other helpful toolkits. With this system we perform ~100-200 DFT calculations per day across various chemistries and materials. A lot of work is put into the active learning system to find and schedule interesting calculations. You can read more about this system
- Dynamic Workflows for Routine Materials Discovery in Surface Science
- Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution
AMPtorch
We are part of a DOE-funded group led by Prof Andy Peterson at Brown to develop machine learning potentials and the AMP code for exascale machines. We have been exploring pytorch as a system for faster and more complicated training systems. We are also working on various active learning strategies to accelerate the train/predict/calculate pipeline.
Graph Convolution Networks for Surface Chemistry
We showed that crystal graph convolution networks can be applied to surface chemistry and catalysis with some modifications. Our fork of the repo has the changes made in our papers. We are in the process of making these models easier to use and develop with collaborators.
- Convolutional Neural Network of Atomic Surface Structures To Predict Binding Energies for High-Throughput Screening of Catalysts
- Toward Predicting Intermetallics Surface Properties with High-Throughput DFT and Convolutional Neural Networks
UQ in Graph Convolution Networks
Uncertainty estimation is important in engineering and active learning, and UQ applied to graph convolution networks is still an emerging area. We explored a number of different strategies and collected our results as a series of jupyter notebooks.
Quantum Espresso in Google Colab workbooks
We use Google colab and other hosted jupyter instances for a lot of our day to day work. We’ve been exploring how to demonstrate active learning strategies with DFT calculations on these systems, and showed that Google Colab GPU instances can actually run QE very quickly! See John Kitchin’s updated DFT book for examples.
Computational Catalysis Datasets
Large consistent datasets can be difficult to find and use in catalysis, and this has limited the development of deep learning models and representations. We’re working with collaborators to improve this further, but for now you can find the datasets we have published or use internally for developing new methods.
Surface chemistry intermediates across intermetallics
We have performed about 100,000 adsorption energy calculations over the past couple years across a range of intermetallics. About 50,000 are considered high quality, with relatively little surface/adsorbate movement. This subset is what we use for training.
- The data for our seminal work can be found in the paper’s repository along with instructions for opening it.
- A more recent dataset can be found here, from the repository of our paper on uncertainty. These data are read the same way the previous dataset is read, except they are opened with
pickleinstead ofjson.
If the function is not working, you may need to downgrade your ASE to version 3.17. If you have any questions or comments about or data, do not hesitate to contact us.
Cleavage energies for intermetallics
Surface energies for asymmetric inorganic surfaces are not well defined, but are important for bimetallic nanoparticle shape and faceting. We calculated several thousand cleavage energies across a range of stable inorganic crystals from the Materials Project, which we used for training graph convolution networks. We are working with collaborators to improve on asymmetric limitations.