Adsorption energy calculations using a pre-trained OCP calculator with OC20 referencing
Contents
Adsorption energy calculations using a pre-trained OCP calculator with OC20 referencing#
Now that we’ve seen how the toy EMT calculator works for the *OCH3 adsorbate (Adsorption energy calculations using a toy calculator), let’s see how a modern OCP calculator does for the same job.
To make this easy, we provide an ASE-compatible calculator calculator to interface with ASE’s functionality. Note that ASE is GPL-licensed!
For this tutorial we download one of the better current best model checkpoint (GemNet-T). This checkpoint has a few important caveats to keep in mind before using it blindly:
This is trained as a force field to predict both energies and forces for specific configurations
The energies predicted are referenced to the bare surface and the same linear combination of species in OC20 (see Adsorption energy calculations using a toy calculator for details on the reference scheme)
The energies and forces are not internally consistent, since we’ve found that models that predict the energy and force separately tend to do better for OC20
Download pretrained checkpoint#
We have released checkpoints of all the models on the leaderboard here. These trained models can be used as an ASE calculator for various calculations.
!wget -q -nc https://dl.fbaipublicfiles.com/opencatalystproject/models/2021_08/s2ef/gemnet_t_direct_h512_all.pt
checkpoint_path = "gemnet_t_direct_h512_all.pt"
Using the OCP Calculator#
import os
import ase.io
import numpy as np
from ase.build import add_adsorbate, fcc100, molecule
from ase.constraints import FixAtoms
from ase.optimize import LBFGS
from ocpmodels.common.relaxation.ase_utils import OCPCalculator
# Construct a sample structure, similar to the EMT relaxation example!
adslab = fcc100("Cu", size=(3, 3, 3))
adsorbate = molecule("CH3O")
add_adsorbate(adslab, adsorbate, 3, offset=(1, 1))
tags = np.zeros(len(adslab))
tags[18:27] = 1
tags[27:] = 2
adslab.set_tags(tags)
cons = FixAtoms(indices=[atom.index for atom in adslab if (atom.tag == 0)])
adslab.set_constraint(cons)
adslab.center(vacuum=13.0, axis=2)
adslab.set_pbc(True)
# Define the calculator
calc = OCPCalculator(checkpoint=checkpoint_path)
# Set up the calculator
adslab.set_calculator(calc)
os.makedirs("data", exist_ok=True)
opt = LBFGS(adslab, trajectory="data/ml_Cu100+CH3O.traj")
opt.run(fmax=0.05, steps=100)
amp: false
cmd:
checkpoint_dir: /home/runner/work/ml_catalysis_tutorials/ml_catalysis_tutorials/ml-catalysis-tutorials/notes/pretrained_models/checkpoints/2022-10-31-18-01-36
commit: cba9fb6
identifier: ''
logs_dir: /home/runner/work/ml_catalysis_tutorials/ml_catalysis_tutorials/ml-catalysis-tutorials/notes/pretrained_models/logs/tensorboard/2022-10-31-18-01-36
print_every: 100
results_dir: /home/runner/work/ml_catalysis_tutorials/ml_catalysis_tutorials/ml-catalysis-tutorials/notes/pretrained_models/results/2022-10-31-18-01-36
seed: null
timestamp_id: 2022-10-31-18-01-36
dataset: null
gpus: 0
logger: tensorboard
model: gemnet_t
model_attributes:
activation: silu
cbf:
name: spherical_harmonics
cutoff: 6.0
direct_forces: true
emb_size_atom: 512
emb_size_bil_trip: 64
emb_size_cbf: 16
emb_size_edge: 512
emb_size_rbf: 16
emb_size_trip: 64
envelope:
exponent: 5
name: polynomial
extensive: true
max_neighbors: 50
num_after_skip: 2
num_atom: 3
num_before_skip: 1
num_blocks: 3
num_concat: 1
num_radial: 128
num_spherical: 7
otf_graph: true
output_init: HeOrthogonal
rbf:
name: gaussian
regress_forces: true
noddp: false
optim:
batch_size: 32
clip_grad_norm: 10
ema_decay: 0.999
energy_coefficient: 1
eval_batch_size: 32
eval_every: 5000
factor: 0.8
force_coefficient: 100
loss_energy: mae
loss_force: l2mae
lr_initial: 0.0005
max_epochs: 80
mode: min
num_workers: 2
optimizer: AdamW
optimizer_params:
amsgrad: true
patience: 3
scheduler: ReduceLROnPlateau
slurm: {}
task:
dataset: trajectory_lmdb
description: Regressing to energies and forces for DFT trajectories from OCP
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
metric: mae
train_on_free_atoms: true
type: regression
trainer: forces
/home/runner/work/ml_catalysis_tutorials/ml_catalysis_tutorials/ocp/ocpmodels/preprocessing/atoms_to_graphs.py:139: UserWarning: Creating a tensor from a list of numpy.ndarrays is extremely slow. Please consider converting the list to a single numpy.ndarray with numpy.array() before converting to a tensor. (Triggered internally at /opt/conda/conda-bld/pytorch_1639180507909/work/torch/csrc/utils/tensor_new.cpp:201.)
cell = torch.Tensor(atoms.get_cell()).view(1, 3, 3)
/home/runner/micromamba-root/envs/buildenv/lib/python3.9/site-packages/torch/autocast_mode.py:141: UserWarning: User provided device_type of 'cuda', but CUDA is not available. Disabling
warnings.warn('User provided device_type of \'cuda\', but CUDA is not available. Disabling')
/home/runner/micromamba-root/envs/buildenv/lib/python3.9/site-packages/torch/functional.py:1069: UserWarning: torch.meshgrid: in an upcoming release, it will be required to pass the indexing argument. (Triggered internally at /opt/conda/conda-bld/pytorch_1639180507909/work/aten/src/ATen/native/TensorShape.cpp:2157.)
return _VF.cartesian_prod(tensors) # type: ignore[attr-defined]
/home/runner/work/ml_catalysis_tutorials/ml_catalysis_tutorials/ocp/ocpmodels/models/gemnet/gemnet.py:373: UserWarning: __floordiv__ is deprecated, and its behavior will change in a future version of pytorch. It currently rounds toward 0 (like the 'trunc' function NOT 'floor'). This results in incorrect rounding for negative values. To keep the current behavior, use torch.div(a, b, rounding_mode='trunc'), or for actual floor division, use torch.div(a, b, rounding_mode='floor').
neighbors_new // 2,
/home/runner/work/ml_catalysis_tutorials/ml_catalysis_tutorials/ocp/ocpmodels/models/gemnet/gemnet.py:467: UserWarning: __floordiv__ is deprecated, and its behavior will change in a future version of pytorch. It currently rounds toward 0 (like the 'trunc' function NOT 'floor'). This results in incorrect rounding for negative values. To keep the current behavior, use torch.div(a, b, rounding_mode='trunc'), or for actual floor division, use torch.div(a, b, rounding_mode='floor').
block_sizes = neighbors // 2
Step Time Energy fmax
LBFGS: 0 18:01:52 0.702756 2.5280
LBFGS: 1 18:01:53 0.610855 0.9909
LBFGS: 2 18:01:53 0.573561 0.5541
LBFGS: 3 18:01:53 0.562686 0.3856
LBFGS: 4 18:01:54 0.547591 0.3904
LBFGS: 5 18:01:54 0.529304 0.4272
LBFGS: 6 18:01:54 0.507553 0.4552
LBFGS: 7 18:01:55 0.479591 0.4565
LBFGS: 8 18:01:55 0.440622 0.6339
LBFGS: 9 18:01:56 0.352820 1.0586
LBFGS: 10 18:01:56 0.161986 1.6880
LBFGS: 11 18:01:56 0.055566 2.5579
LBFGS: 12 18:01:57 0.043263 1.5631
LBFGS: 13 18:01:57 -0.021224 1.3076
LBFGS: 14 18:01:58 -0.255827 1.2519
LBFGS: 15 18:01:58 -0.284720 0.8289
LBFGS: 16 18:01:58 -0.331042 0.7366
LBFGS: 17 18:01:59 -0.422400 0.9306
LBFGS: 18 18:01:59 -0.454963 0.6932
LBFGS: 19 18:02:00 -0.486615 0.4053
LBFGS: 20 18:02:00 -0.495915 0.3343
LBFGS: 21 18:02:00 -0.517762 0.4154
LBFGS: 22 18:02:01 -0.529808 0.4467
LBFGS: 23 18:02:01 -0.546710 0.3477
LBFGS: 24 18:02:02 -0.553586 0.3163
LBFGS: 25 18:02:02 -0.558584 0.3828
LBFGS: 26 18:02:03 -0.557754 0.4700
LBFGS: 27 18:02:03 -0.550925 0.6269
LBFGS: 28 18:02:03 -0.552629 0.5928
LBFGS: 29 18:02:04 -0.569216 0.3315
LBFGS: 30 18:02:04 -0.582791 0.2663
LBFGS: 31 18:02:05 -0.591911 0.2639
LBFGS: 32 18:02:05 -0.599708 0.2363
LBFGS: 33 18:02:05 -0.604322 0.2343
LBFGS: 34 18:02:06 -0.606322 0.2199
LBFGS: 35 18:02:06 -0.608697 0.3313
LBFGS: 36 18:02:07 -0.612839 0.4964
LBFGS: 37 18:02:07 -0.626581 0.6773
LBFGS: 38 18:02:07 -0.654602 1.0733
LBFGS: 39 18:02:08 -0.732619 0.8797
LBFGS: 40 18:02:08 -0.793629 1.0103
LBFGS: 41 18:02:09 -0.821329 1.3485
LBFGS: 42 18:02:09 -0.895005 0.7361
LBFGS: 43 18:02:09 -0.931382 0.4471
LBFGS: 44 18:02:10 -0.962967 0.4605
LBFGS: 45 18:02:10 -1.002134 0.4616
LBFGS: 46 18:02:11 -1.028943 0.4869
LBFGS: 47 18:02:11 -1.072585 0.6643
LBFGS: 48 18:02:11 -1.093312 0.6843
LBFGS: 49 18:02:12 -1.130317 0.4795
LBFGS: 50 18:02:12 -1.146077 0.3814
LBFGS: 51 18:02:13 -1.175920 0.5125
LBFGS: 52 18:02:13 -1.198479 0.4924
LBFGS: 53 18:02:13 -1.217808 0.4121
LBFGS: 54 18:02:14 -1.232873 0.2768
LBFGS: 55 18:02:14 -1.237812 0.3241
LBFGS: 56 18:02:15 -1.241352 0.2208
LBFGS: 57 18:02:15 -1.251641 0.2282
LBFGS: 58 18:02:15 -1.266796 0.2116
LBFGS: 59 18:02:16 -1.273413 0.1641
LBFGS: 60 18:02:16 -1.277564 0.1170
LBFGS: 61 18:02:17 -1.279324 0.1176
LBFGS: 62 18:02:17 -1.282491 0.1352
LBFGS: 63 18:02:18 -1.287317 0.1897
LBFGS: 64 18:02:18 -1.291918 0.2052
LBFGS: 65 18:02:18 -1.293563 0.1642
LBFGS: 66 18:02:19 -1.300365 0.1217
LBFGS: 67 18:02:19 -1.302982 0.1184
LBFGS: 68 18:02:20 -1.303218 0.1577
LBFGS: 69 18:02:20 -1.305581 0.1885
LBFGS: 70 18:02:20 -1.308966 0.1878
LBFGS: 71 18:02:21 -1.312502 0.1661
LBFGS: 72 18:02:21 -1.314188 0.1247
LBFGS: 73 18:02:22 -1.316728 0.1075
LBFGS: 74 18:02:22 -1.317776 0.1017
LBFGS: 75 18:02:22 -1.316023 0.1204
LBFGS: 76 18:02:23 -1.314013 0.1141
LBFGS: 77 18:02:23 -1.316531 0.1486
LBFGS: 78 18:02:24 -1.319495 0.1470
LBFGS: 79 18:02:24 -1.323242 0.1332
LBFGS: 80 18:02:24 -1.329728 0.1107
LBFGS: 81 18:02:25 -1.334422 0.1026
LBFGS: 82 18:02:25 -1.342553 0.0958
LBFGS: 83 18:02:26 -1.348809 0.0705
LBFGS: 84 18:02:26 -1.354896 0.0399
True
Now, let’s visualize the output!
import matplotlib.pyplot as plt
from ase.visualize.plot import animate
from matplotlib import rc
# the `index` argument corresponds to what frame of the trajectory to read in, specifiying ":" reads in the full trajectory.
traj = ase.io.read("data/ml_Cu100+CH3O.traj", index=":")
anim = animate(traj, radii=0.8, rotation=("-75x, 45y, 10z"))
rc("animation", html="jshtml")
anim
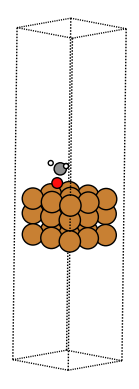
Compare the results here with what EMT predicted in Adsorption energy calculations using a toy calculator. Note that the adsorbate rotates so that the O is pointing down on the surface and the adsorbate does not associate. This looks much more reasonable!
We can plot the adsorption energy directly since this OCP calculator directly outputs referenced energies. We don’t need to repeat the gas-phase and bare slab calculations like we did in Adsorption energy calculations using a toy calculator.
import pandas as pd
import plotly.express as px
energies = [image.get_potential_energy() for image in traj]
df = pd.DataFrame(
{
"Relaxation Step": range(len(energies)),
"GemNet-T Adsorption Energy [eV]": energies,
}
)
fig = px.line(df, x="Relaxation Step", y="GemNet-T Adsorption Energy [eV]")
fig.show()